
The Gut Connection: A New Chapter in Metformin’s Story
How new science explains what metformin does in your gut, and why it’s safer than you may think

Metformin is one of the most prescribed drugs in the world. It has been in continuous clinical use for decades, has an outstanding safety record, and remains the first-line treatment for type 2 diabetes in virtually every major clinical guideline. It is also a drug whose mechanism of action was, until very recently, incompletely understood.
Two papers published last week, both from Navdeep Chandel’s research group at Northwestern University, add a significant piece to that puzzle.
What We Already Knew
Metformin works through several distinct mechanisms simultaneously, which is part of why it is so effective.
Hepatic gluconeogenesis suppression. The liver continuously manufactures new glucose from non-sugar precursors, a process called gluconeogenesis. According to a 2023 review in Nature Reviews Endocrinology by Foretz, Guigas, and Viollet, suppression of hepatic glucose production has long been considered the drug’s primary mechanism of action.
AMPK activation. Metformin activates an enzyme called AMPK, which functions as a cellular fuel gauge: it shifts cells away from energy-consuming processes and toward energy conservation and glucose uptake. In simple terms: it makes cells more responsive to glucose already circulating in the bloodstream. As reviewed by LaMoia and Shulman in Endocrine Reviews (2021), AMPK activation contributes to metformin’s insulin-sensitizing effects across multiple tissues.
Gut microbiome modulation. Metformin alters the composition of the bacterial ecosystem in the gut in ways that appear beneficial for glucose metabolism. The mechanisms connecting microbiome changes to glycemic control are still being worked out, but the association is well established in human data (Foretz et al., 2023).
GLP-1 secretion. Metformin increases the release of GLP-1, a gut hormone that stimulates insulin secretion in response to meals and slows gastric emptying. This contributes to the drug’s lowering effect on after-meal blood glucose levels (Foretz et al., 2023).
A Pharmacokinetic Problem That Needed Solving
There was always something uncomfortable about the hepatocentric model, and researchers knew it.
Inhibiting complex I, the first and largest component of the mitochondrial electron transport chain and a key step in cellular energy production, requires metformin concentrations in the millimolar range when tested in isolated cell systems. That concentration is simply not what standard oral doses deliver to the liver or to the bloodstream.
Sebo et al. (2026), published in Nature Metabolism, measured tissue concentrations directly in the experimental animals used in their study and found what prior human pharmacokinetic literature had suggested: intestinal epithelial concentrations of metformin exceed plasma concentrations by up to 300-fold, and hepatic concentrations by 10 to 100-fold. The gut, specifically the lining of the small intestine, is where metformin concentrates most heavily after an oral dose, and where the drug reaches the concentrations required to inhibit complex I.
The New Evidence
To test whether complex I inhibition specifically in intestinal cells is necessary for metformin to work, the researchers created mice whose intestinal cells were genetically engineered to express a yeast enzyme called NDI1. This enzyme bypasses mammalian complex I entirely and is completely resistant to metformin’s inhibitory effect. In these mice, the drug can still circulate and reach other tissues, but the intestinal cells simply ignore its effect on their energy machinery.
The first paper, Reczek et al. (2026) in Science Advances, used mice with the NDI1 bypass expressed throughout the whole body. Metformin’s glucose-lowering effect was significantly attenuated compared to normal controls, under both standard and high-fat diet conditions. Tissue drug concentrations were equivalent in both groups, ruling out the possibility that the engineered mice were simply absorbing or distributing the drug differently.
The second paper, Sebo et al. (2026) in Nature Metabolism, used a more precise model: NDI1 expressed only in intestinal epithelial cells, using a targeting system called Villin-Cre. In these intestine-specific mice, metformin-induced glucose uptake by the intestine was abolished. Citrulline suppression was attenuated, as was GDF15 elevation, lactate production, and postprandial glucose lowering. Multiple independent readouts pointed in the same direction.
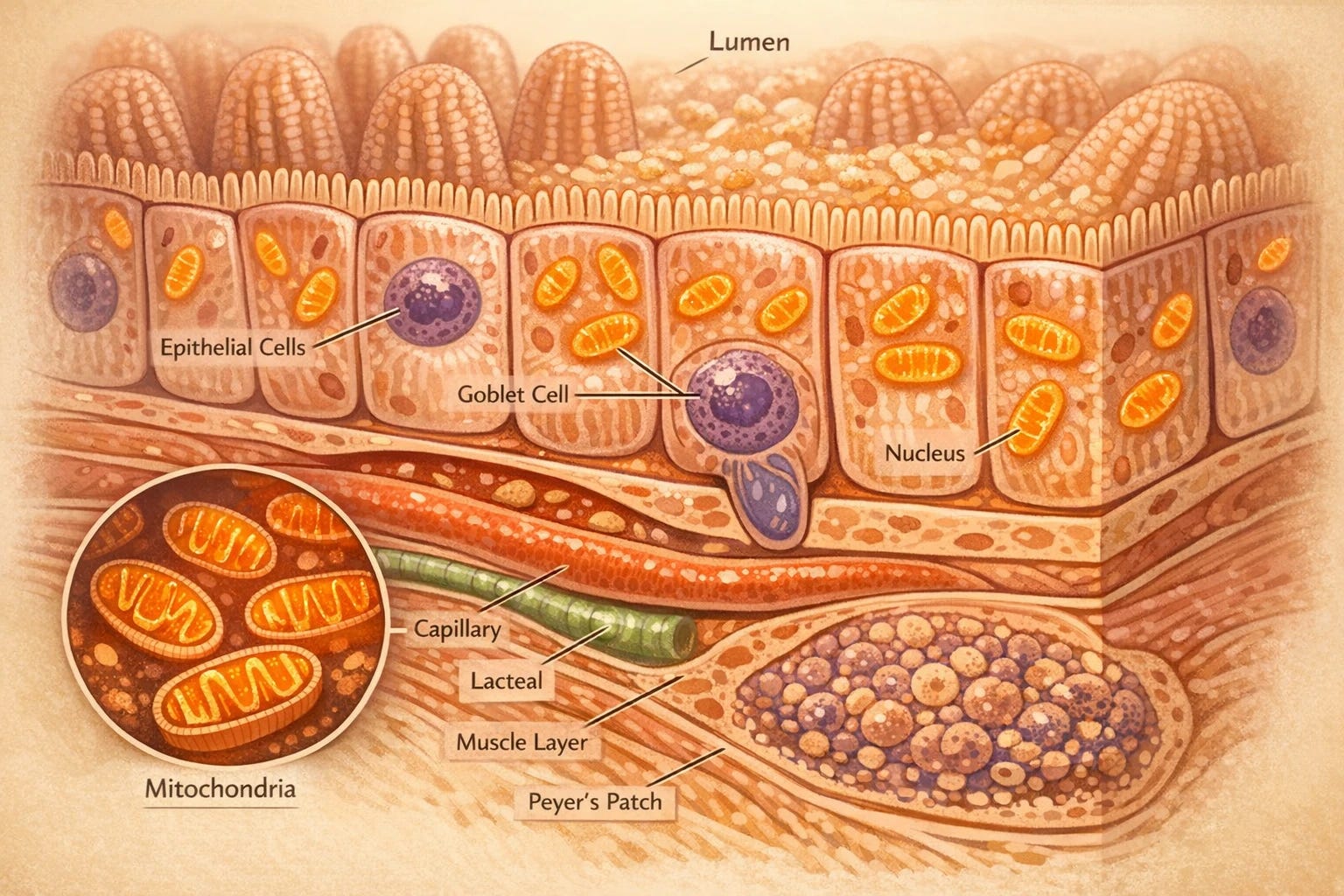
The mechanism works as follows. When metformin inhibits complex I in intestinal epithelial cells, those cells can no longer produce energy through their normal oxidative pathway efficiently. They compensate by shifting to an alternative strategy, metabolizing glucose through glycolysis instead. The gut, in effect, becomes a glucose sink: it intercepts a portion of the glucose arriving from a meal and use it before that glucose can reach the bloodstream in full. The intestine is doing metabolic work that was previously attributed primarily to the liver.
These are animal model findings. Mouse physiology is not human physiology, and the NDI1 bypass was incomplete, meaning the engineered mice still showed partial responses to the drug. This tells us that additional mechanisms are at work even in the mouse model. What proportion of metformin’s effect in humans operates through this intestinal mechanism will require dedicated human validation studies before we can answer with confidence.
The Citrulline Biomarker
Citrulline is an amino acid synthesized almost exclusively in the mitochondria of small intestinal cells. The enzymes that make it, CPS1 and OTC, depend on mitochondrial ATP. When complex I is inhibited and mitochondrial ATP output drops, citrulline synthesis falls. The circulating citrulline in your bloodstream is, in practical terms, a readout of what the mitochondria in your intestinal lining are doing.
Sebo et al. (2026) drew on two independent human datasets to test whether metformin produces the expected citrulline drop in people. The first, from Rotroff et al. published in Frontiers in Pharmacology (2016), involved 33 non-diabetic subjects. The second, from Aleidi et al. in Frontiers in Pharmacology (2021), involved 26 patients with obesity and type 2 diabetes. In both cohorts, citrulline was the most significantly downregulated metabolite following metformin treatment.
This human data does not prove that the intestinal mechanism drives glycemic control in people the way the mouse experiments demonstrate in animals. But it is consistent across two independent cohorts, and consistent with prior reports from other groups.
If you enjoy evidence-based medical information, subscribe to receive these articles delivered to your mailbox every week.
What This Explains About Metformin in Practice
The gut mechanism, once you understand it, makes sense of several clinical observations that were previously explained incompletely or not at all.
Postprandial glucose control. Metformin is particularly effective at blunting the spike in blood glucose that follows a meal. Under the hepatocentric model, this was somewhat difficult to account for precisely. Under the gut-centric model, it is exactly what you would predict: the intestine is intercepting glucose at the point of absorption, buffering the postprandial load before it reaches the portal circulation.
Why mealtime dosing matters. Clinical guidance has long recommended taking metformin with food, primarily to reduce gastrointestinal side effects. The new data suggests a second, reason: the drug needs to be present in the intestinal lumen when glucose is arriving from a meal to function as a glucose buffer. Sebo et al. (2026) found that administering metformin continuously in drinking water, which produces chronic low-level exposure rather than mealtime boluses, failed to achieve glycemic control in the mouse model.
GI side effects. Nausea, bloating, and diarrhea are the most common reasons patients discontinue metformin, particularly early in treatment. Under the gut-centric model this is a direct consequence of where the primary action is occurring. The intestinal epithelium is adapting to a meaningful disruption in its energy metabolism. The clinical implication is that these side effects can be substantially minimized by increasing the dose slowly over weeks to months, allowing the gut time to adapt. This is consistent with what gradual titration reliably produces in practice.
GDF15 elevation. GDF15 is a stress hormone released by cells under metabolic pressure. Metformin raises GDF15 levels, and a 2020 paper in Nature by Coll et al. demonstrated that GDF15 mediates metformin’s effects on body weight and appetite suppression. The intestinal complex I inhibition model provides an explanation for why the gut would generate this signal: the cells are under energy stress, and GDF15 is part of their stress response.
There is evidence that metformin may blunt some of the mitochondrial adaptations to aerobic exercise in older adults, as reported in studies by Konopka et al. and Walton et al. in Aging Cell (2019). This does not change the risk-benefit for most patients with type 2 diabetes, but it is worth knowing, particularly for patients who are simultaneously pursuing resistance or aerobic training programs. The conversation belongs between patient and physician, with full awareness of the data.
Putting the Lactic Acidosis Risk in Perspective
If you have been on metformin for any length of time, you may have heard or read about lactic acidosis. It is the complication most frequently cited by people who are anxious about the drug.
Lactic acidosis is a serious condition in which lactic acid accumulates in the blood faster than the body can clear it. It carries significant mortality when it occurs. The anxiety around metformin and lactic acidosis, however, is mostly inherited from a different drug.

Phenformin was a biguanide, the same drug family as metformin, used for type 2 diabetes until 1977, when it was withdrawn from the US market because of an unacceptable rate of lactic acidosis: approximately 40 to 64 cases per 100,000 patient-years (Foretz et al., 2023).
Metformin-associated lactic acidosis in appropriately selected patients occurs at an estimated rate of 3 to 10 cases per 100,000 patient-years (Foretz et al., 2023), roughly one-tenth of the phenformin rate, and almost invariably in the context of significant confounders: severe renal failure, liver disease, hemodynamic instability, or situations where lactate clearance is already compromised for independent reasons.
The May 2026 papers provide an explanation for why these two drugs in the same chemical family carry such different risk profiles. The IC50 values, the concentration required to inhibit half of complex I activity, tell the story. Metformin’s IC50 is approximately 19,400 micromolar. Phenformin’s is approximately 430 micromolar (Bridges et al., Biochemical Journal, 2014; Turner et al., Diabetes, 2008, as cited in Sebo et al., 2026). Metformin requires roughly 45 times the concentration of phenformin to achieve the same degree of complex I inhibition.
In practice, metformin concentrates in the intestinal epithelium and produces its therapeutic effect there. It does not reach systemic complex I inhibition at standard therapeutic doses in patients with normal renal function. Phenformin, far more potent, produces systemic complex I inhibition more readily, leading to excess lactate production across multiple tissues.
In 40 years of practice, I encountered one single case of suspected metformin-associated lactic acidosis. The patient had significant renal failure, and in hemodynamic shock following acute blood loss from a traffic accident; two well known causes of metabolic acidosis. Attribution to metformin alone would have been a substantial inferential stretch here.
Metformin should be used with care or avoided in severe renal impairment, advanced liver disease, and acute illness with hemodynamic compromise. Within those boundaries, it has an outstanding safety record.
Berberine Mechanism Confirmed
Berberine is a plant-derived compound marketed as a “natural” alternative to metformin, and the “nature’s Ozempic” label has circulated widely in wellness spaces. These papers confirm that berberine does, in fact, work through the same intestinal complex I mechanism as metformin. In the intestine-specific NDI1 mouse model, berberine’s glucose-lowering effect was completely abolished, similar to metformin’s results.
The IC50 data is worth considering here. Berberine inhibits complex I at a concentration of approximately 15 micromolar. Phenformin, the drug withdrawn from the US market for causing fatal lactic acidosis at scale, requires approximately 430 micromolar. Metformin requires approximately 19,400 micromolar. Berberine is roughly 30 times more potent a complex I inhibitor than phenformin, and approximately 1,300 times more potent than metformin (Bridges et al., 2014; Turner et al., 2008, as cited in Sebo et al., 2026).
Berberine appears to be safe in practice, the reason is likely poor intestinal absorption. Berberine is poorly absorbed from the gut in standard supplement doses, and it is actively pumped back into the intestinal lumen by a transporter called P-glycoprotein. The drug is essentially trapped in the intestine limiting its systemic absorption.
Plants evolved berberine as a chemical defense against the insects and animals that fed on them, its target being the mitochondrial energy-generating machinery inside cells. The human gut developed the P-glycoprotein system as a defense mechanism that actively pumps berberine back into the gut before it can reach the bloodstream in significant quantities.
Berberine is also a P-glycoprotein inhibitor: it can block the very pump that keeps it gut-restricted. Therefore is theoretically possible, that at higher doses berberine may progressively undermine its own containment mechanism leading to higher risk for lactic acidosis.
Sebo et al. (2026) tested this directly. Co-administering berberine with encequidar, an intestine-specific P-glycoprotein inhibitor, dramatically amplified berberine’s glucose-lowering effect in normal mice and had no effect in the intestine-specific NDI1 mice, confirming that the amplification worked by increasing intestinal complex I inhibition.
A number of commonly used medications inhibit the P-glycoprotein mechanism including grapefruit, verapamil, diltiazem, amiodarone, carvedilol, quinidine, dronedarone, clarithromycin (and other macrolide antibiotics), cyclosporine, ritonavir, itraconazole and ketoconazole. This interaction is unmonitored in supplement users, is not disclosed on berberine labels, and is entirely outside the safety oversight that applies to regulated pharmaceuticals.
There are no confirmed human case reports of berberine-induced lactic acidosis in the published literature. But the same mechanism that explains phenformin’s withdrawal from the market raises a coherent theoretical concern about berberine in patients on P-glycoprotein-inhibiting medications.
There is no rational clinical argument for choosing berberine over metformin in a patient who is a candidate for pharmacological treatment of type 2 diabetes or prediabetes. Metformin has decades of rigorous human trial data behind it, a known and manageable safety profile, regulatory oversight, quality-controlled manufacturing, and defined contraindications.
Berberine, being an unregulated over-the-counter supplement, has none of those things.
What This Means if You Are on Metformin
If you are currently taking metformin, nothing about the May 2026 findings changes your treatment. These papers add new scientific explanations of why it works.
The gut-centric model reinforces two practices that clinical guidance has long recommended and that are now understood to be important rather than merely convenient: take metformin with meals, and if you are starting the drug or increasing the dose, do so gradually. The intestine is where the action is, and it benefits from time to adapt.
If you have been avoiding metformin out of concern about lactic acidosis, the risk data and the information above are worth discussing with your physician in the context of your specific situation. Fear inherited from phenformin’s history, applied without adjustment to metformin’s actual pharmacology, has likely led patients to avoid a drug that would have served them well.
And if you have been taking berberine as an alternative to a conversation with your physician about metformin, this is a good moment to have that conversation.
Found this article useful? Share your thoughts. Join the conversation below.
Educational content on The Metabolic Archives is free, because medical information should be accessible to everyone. If you find value and want to support the work, a paid subscription is available and genuinely appreciated. Visit the About Page for additional information.
The Metabolic Archives is for educational and informational purposes only, and is not intended as medical advice, diagnosis, or treatment, and does not constitute a doctor-patient relationship. Do not adopt any recommendation discussed in any article or guides published here, make changes or abandon any prescribed medical treatment without prior consultation with your physician. Always seek the advice of your physician or other qualified health provider for any questions regarding your medical condition and recommended treatment options.
By reading this post, you acknowledge that you have read and agree to the Terms of Service of The Metabolic Archives, which govern all use of this content including restrictions on reproduction.
© 2026 The Metabolic Archives. All rights reserved.
I’ve read articles that suggest metformin may help with inflammatory arthritis. Do you have an opinion about this?