
The Root System: A Complete Guide to Insulin Resistance
Part Four: When Every System Pays the Price

Part Three established insulin resistance as a single systemic failure manifesting across multiple organ systems. The conditions covered there — type 2 diabetes, dyslipidemia, obesity, PCOS, hypertension — share the same roots.
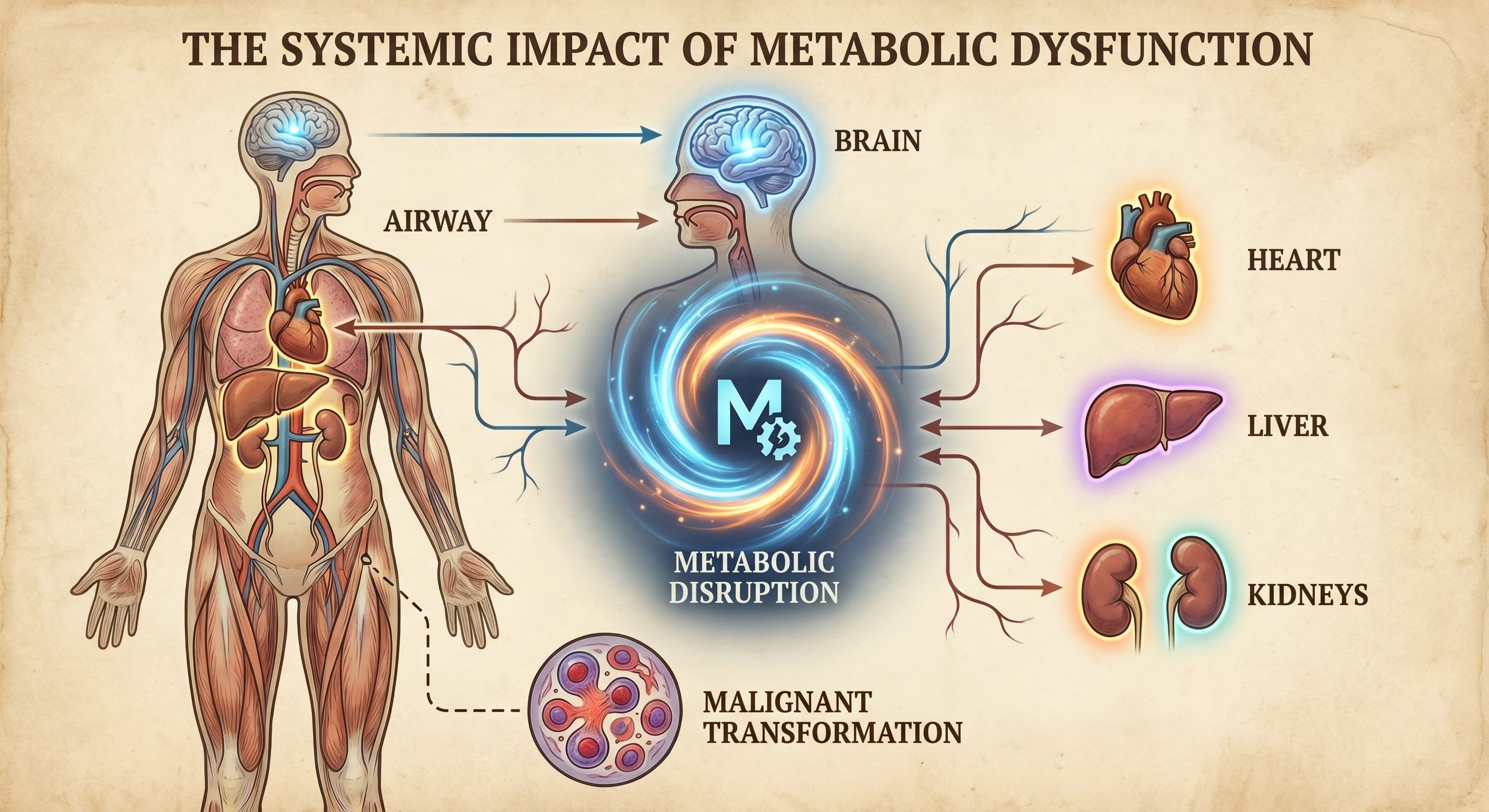
So do the six that follow.
Cardiovascular Disease
Cardiovascular disease is the leading cause of death in the USA. Five mechanisms are at the center of this process:
First, insulin resistance in the vascular endothelium impairs nitric oxide production through the PI3K-eNOS pathway. Nitric oxide is the primary vasodilatory and anti-atherogenic signal in the arterial wall. The endothelium becomes adhesive, permeable, and pro-inflammatory before any visible lesion exists.
Second, the atherogenic dyslipidemic triad — elevated triglycerides, reduced HDL, small dense LDL predominance — facilitate accelerated plaque substrate. Small dense LDL particles are more susceptible to oxidation and are retained longer in the subendothelial space than large buoyant LDL. Standard lipid panels miss this pattern entirely.
Third, the chronic systemic inflammation that both drives and is driven by insulin resistance promotes plaque instability. Vulnerable plaques rupture, stable ones do not.
Fourth, elevated plasminogen activator inhibitor-1 (PAI-1) in insulin resistant populations impairs fibrinolysis, shifting the thrombotic balance toward clot formation (Alessi and Juhan-Vague, Arteriosclerosis, Thrombosis, and Vascular Biology, 2006). A plaque that ruptures in a patient with normal fibrinolytic capacity may produce a transient event. In a patient with PAI-1 elevation, the same rupture is more likely to produce vessel occlusion.
Fifth, direct cardiac insulin resistance impairs myocardial glucose utilization and reduces ischemic resilience; the heart becomes metabolically inefficient before coronary disease produces ischemia (Riehle and Abel, Circulation Research, 2016).
These five mechanisms operate concurrently in the same arterial wall.
Metabolic disease is a powerful driver of cardiovascular disease through multiple mechanisms. While aggressive lifestyle interventions are needed to address the underlying insulin resistance, consult with your medical team BEFORE starting any exercise program to verify is safe for you.
Chronic Kidney Disease (CKD)
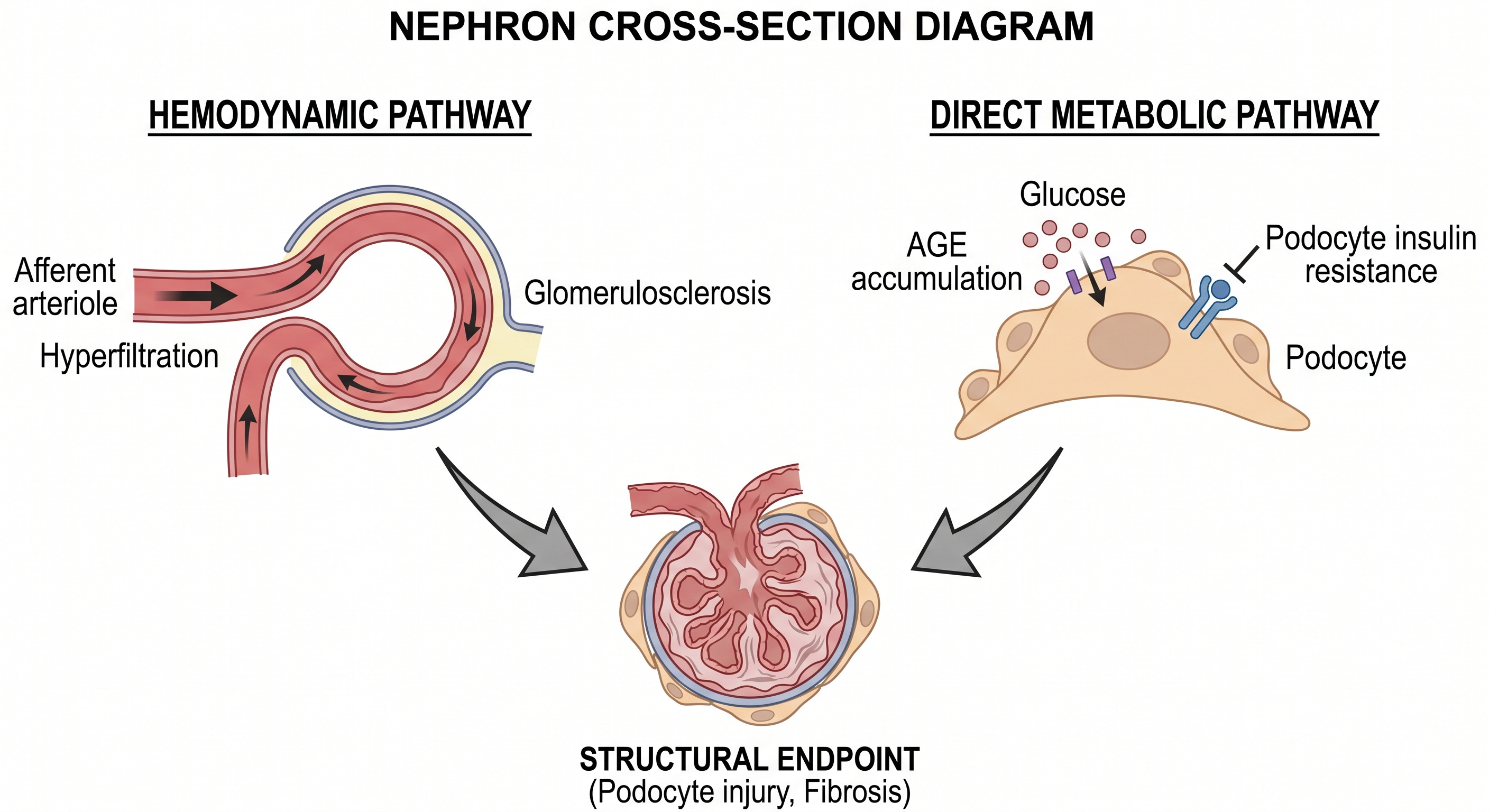
The kidney sustains damage through two parallel and independent pathways, and then amplifies the metabolic disruption that caused it.
The hemodynamic pathway operates through hypertension-driven glomerular hyperfiltration and the renin-angiotensin-aldosterone system (RAAS)-mediated glomerulosclerosis. Chronically elevated intraglomerular pressure damages the filtration architecture over years, producing the progressive proteinuria and glomerular filtration rate (GFR) decline that define CKD’s clinical course.
The metabolic pathway operates independently of blood pressure. Advanced glycation end-products accumulate in the glomerular basement membrane, impairing its structural integrity. Podocytes, the specialized cells that form the filtration barrier’s final layer, require intact insulin signaling to maintain their architecture. Podocyte insulin resistance impairs that architecture directly, producing proteinuria through a mechanism that antihypertensive therapy does not address (Welsh et al., Cell Metabolism, 2010).
Renal disease comes as a consequence of dual mechanisms: elevated arterial blood pressure causing direct damage to the filtration units, and insulin resistance impairing the glomerular filtration unit. While controlling arterial blood pressure is an important intervention, particularly with ACEI/ARB class drugs, lifestyle modification is essential to address the underlying metabolic disruption responsible for sustained renal damage.
If you enjoy evidence-based medical information, subscribe to receive these articles delivered to your mailbox every week.
Non-Alcoholic Fatty Liver Disease (NAFLD)
NAFLD is the most anatomically direct consequence of hepatic insulin resistance on this list. The sequence from insulin resistance to hepatocellular carcinoma is defined at each step.
Visceral adipose tissue delivers free fatty acids into the portal circulation in volumes proportional to the adipose mass and insulin resistance severity. Hepatic insulin resistance simultaneously impairs the liver’s capacity to dispose of this lipid load through normal oxidative pathways. De novo lipogenesis adds to the substrate burden. VLDL secretion, insufficient to export the accumulating fat, leaves triglycerides depositing in hepatocytes leading to steatosis (fatty liver).
The transition from steatosis to non-alcoholic steatohepatitis (localized liver inflammation) is the critical inflection point. The same pro-inflammatory cytokine milieu driving peripheral insulin resistance — TNF-alpha, IL-6, and related mediators — activates Kupffer cells and hepatic stellate cells locally. Hepatic inflammation and progressive fibrosis (progressive scar tissue development) follow. Globally, NAFLD is estimated to affect approximately 25% of the world population, a prevalence that tracks the insulin resistance epidemic with precision (Younossi et al., Hepatology, 2016).
NAFLD progresses to cirrhosis through continued fibrotic remodeling, and cirrhosis carries a well-characterized risk of hepatocellular cancer, connecting NAFLD directly to the cancer section that follows.
Routinely described to patients as “a little fat on the liver”, steatosis is the initial finding eventually leading to localized liver tissue inflammation, cirrhosis and increased risk of liver cancer in the future. Fatty liver, although a benign finding, is a major red flag that should prompt adoption of aggressive lifestyle modification.
Obstructive Sleep Apnea (OSA)
Visceral obesity and the fat deposition that accompanies it reduce pharyngeal caliber and elevate the diaphragm, compromising functional residual respiratory capacity in the supine position. The airway that collapses during sleep is narrowed before the patient lies down.
Intermittent hypoxia activates the hypothalamic-pituitary-adrenal axis and sympathetic nervous system, generating cortisol and catecholamine surges that directly impair insulin signaling (Punjabi and Polotsky, Journal of Applied Physiology, 2005). Hypoxia-reoxygenation cycling produces oxidative stress that compounds the signaling impairment. Sleep fragmentation disrupts slow-wave sleep, the phase during which growth hormone secretion and nocturnal glucose regulation are concentrated.
Common diagnostic symptoms of OSA include:
Excessive daytime sleepiness
Loud, chronic snoring
Interrupted breathing while asleep (apnea)
Nocturnal gasping or choking
Un-refreshing sleep (feeling tired after waking up)
Frequent nocturnal awakenings
Nocturia
Morning headache
Cognitive impairment / difficulty concentrating
Irritability or mood disturbance
Decreased libido
Dry mouth or sore throat on waking
Insulin resistance mediated obesity is a mayor cause of OSA, and once OSA is present leads to endocrine dysfunction through stress induced cortisol elevation and oxidative stress that in turn worsens insulin resistance. If you have some of the symptoms listed above is time to seek urgent testing. Untreated OSA is associated with increase the risk of cardiac arrhythmias (particularly atrial fibrillation) and sudden cardiac death, among other complications.
Dementia
Insulin receptors are distributed throughout the central nervous system — densely expressed in the hippocampus, cortex, and hypothalamus — where they regulate synaptic plasticity, neuronal survival, and critical aspects of protein processing (Havrankova et al., Nature, 1978). Brain insulin signaling governs tau phosphorylation and amyloid precursor protein processing through pathways that, when impaired, generate the hallmark pathological features of Alzheimer’s disease (Craft and Watson, Lancet Neurology, 2004).
The “Type 3 diabetes” hypothesis proposes that Alzheimer’s disease represents, at least in part, insulin resistance localized to neural tissue. This framing is supported by postmortem brain insulin signaling data showing impaired IRS-1 and Akt activity in Alzheimer’s brains. It is not universally accepted as a formal diagnostic category, and prospective interventional causation in humans has not been established. The bridge to Alzheimer’s pathology runs through GSK-3 beta hyperactivation driven by Akt underactivity, producing tau hyperphosphorylation and impaired amyloid precursor protein processing (de la Monte and Wands, Journal of Diabetes Science and Technology, 2008).
In simple terms: the brain needs insulin to function properly. In Alzheimer’s some believe the brain loses its ability to respond to insulin. When insulin signaling breaks down in brain cells, it triggers a chain of events that leads to the toxic protein buildups — tangles and plaques — that are the hallmarks of Alzheimer’s disease. This connection is still being actively studied.
Vascular dementia represents a parallel pathway from the same metabolic root. The cerebrovascular consequences of insulin resistance — endothelial dysfunction, hypertension-driven small vessel disease, atherosclerotic large vessel disease — produce ischemic lesions that impair cognition through a mechanism entirely distinct from amyloid pathology. In clinical practice, mixed dementia combining both pathways is common.
The epidemiological picture is not subtle. Type 2 diabetes is associated with approximately doubled risk of Alzheimer’s disease in prospective cohort studies, and midlife metabolic risk factors predict late-life cognitive decline decades in advance (Biessels et al., Lancet Neurology, 2006).
Take away: The implication here is that metabolic health in your forties and fifties may lead to dementia in your senior years by several theoretical mechanisms, something to think about.
Cancer
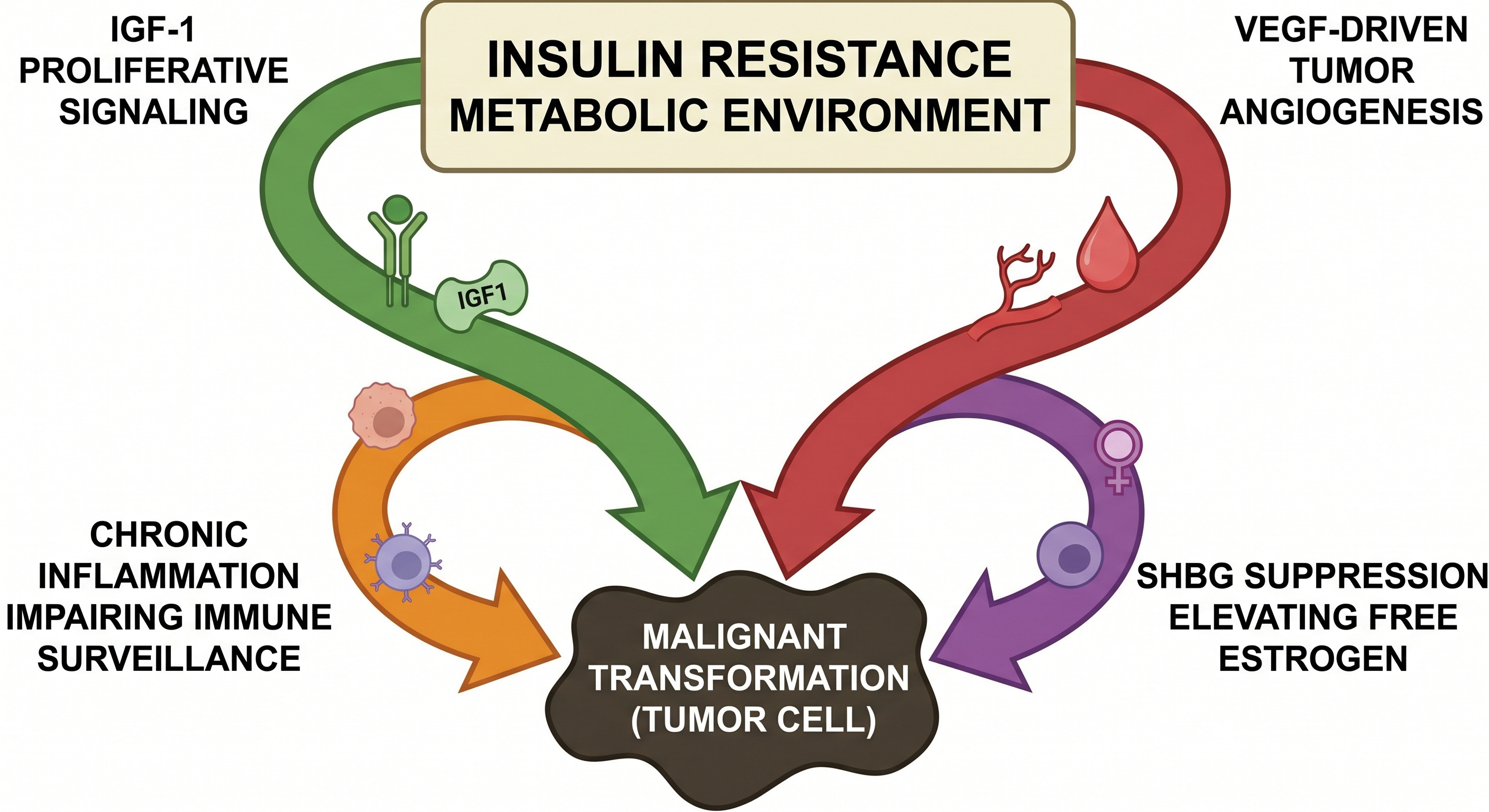
The epidemiological associations between insulin resistance, obesity, and specific malignancies are robust. A systematic review and meta-analysis of prospective observational studies found elevated incidence of colorectal, breast, endometrial, pancreatic, hepatocellular, esophageal, and renal cell carcinomas in obese populations, with dose-dependent relationships in several cancer types (Renehan et al., Lancet, 2008). The hypotheses are compelling and experimentally supported in cellular and animal models. Prospective human interventional causation, the standard confirming insulin resistance as a modifiable cancer risk factor, is not yet fully established.
Four mechanisms lend support to the hypothesis:
Chronic hyperinsulinemia activates insulin and IGF-1 receptors on susceptible cells, engaging the PI3K-Akt-mTOR proliferative cascade while simultaneously suppressing IGFBP-1 and IGFBP-3, the binding proteins that limit free IGF-1 bioavailability. The result is amplified mitogenic and anti-apoptotic signaling in tissues already exposed to a permissive growth environment (Pollak et al., Nature Reviews Cancer, 2004).
In simple terms this favors tumor growth via unrestricted cell division and reduction in programmed cancer cell death.
Insulin and IGF-1 stimulate vascular endothelial growth factor production in tumor-adjacent tissue, supporting the neovascularization that growing tumors require to sustain themselves beyond the diffusion limit of oxygen and nutrients (Warren et al., Journal of Biological Chemistry, 1996). A tumor that cannot recruit its own blood supply cannot grow beyond microscopic size.
Chronic systemic inflammation, the shared metabolic environment of insulin resistance, impairs immune surveillance through mechanisms that are actively under investigation.
Finally, the SHBG suppression produced by hyperinsulinemia elevates free estrogen bioavailability in hormone-sensitive tissues, a mechanism with direct relevance to breast and endometrial malignancy risk.
Observational data show metformin use in diabetic populations is associated with reduced cancer incidence (Evans et al., BMJ, 2005). This is a consistent finding across multiple studies.
For now these are observational findings fueling multiple hypothesis, prospective evidence is not yet available to confirm a concrete cause-effect relationship. This is one of the most actively evolving areas in metabolic medicine research today.
One Disruption. Twelve Conditions.
The human body is an inter-connected biological network, not a collection of independent systems. Disrupt one signaling system and the consequences propagate across multiple organ systems as evidenced here.
Part Five turns from the map to the intervention. What addressing insulin resistance at the root actually requires.
Found this article useful? Share your thoughts. Join the conversation below.
Educational content on The Metabolic Archives is free, because medical information should be accessible to everyone. If you find value and want to support the work, a paid subscription is available and genuinely appreciated. Visit the About Page for additional information.
The Metabolic Archives is for educational and informational purposes only, and is not intended as medical advice, diagnosis, or treatment, and does not constitute a doctor-patient relationship. Do not adopt any recommendation discussed in any article or guides published here, make changes or abandon any prescribed medical treatment without prior consultation with your physician. Always seek the advice of your physician or other qualified health provider for any questions regarding your medical condition and recommended treatment options.
By reading this post, you acknowledge that you have read and agree to the Terms of Service of The Metabolic Archives, which govern all use of this content including restrictions on reproduction.
© 2026 The Metabolic Archives. All rights reserved.